Speed comparisons
speed.RmdHere, I compared the performance of rtrees (v1.0.0) and
V.PhyloMaker (v0.1.0) (V.PhyloMaker2 has
almost the same speed as V.PhyloMaker, so no comparisons
here). In most cases, rtrees is at least two times faster
than V.PhyloMaker (Fig. below). For example, with 50
missing species to bind, the average time used by rtrees is
0.914 second while V.PhyloMaker took 13.6 seconds on
average; with 5,000 missing species to bind, rtrees used
80.9 seconds on average while V.PhyloMaker used 187
seconds. In general, the time used by rtrees and
V.PhyloMaker increased 15.6 and 34.8 seconds on average
with 1,000 more missing species to be grafted, respectively. All tests
were conducted on a 14’ Macbook Pro with M1 pro chip.
Some may not think it is a big difference. However, for me as a user, waiting for more than 10 seconds to have about 50 species grafted is not ideal. Also the accumulated time can be large if one is doing this multiple times (e.g., doing some simulations that require thousands of iterations).
The R code used for the speed tests can be found here.
library(ggplot2)
speed_out = readRDS(system.file("extdata", "rtrees_speed_out.rds", package = "rtrees"))
speed_out = dplyr::mutate(speed_out, n_sp_missing_k = n_sp_missing / 1000, time_s = time/1e9)
# speed_lm = dplyr::group_by(speed_out, expr) |>
# dplyr::do(broom::tidy(lm(time_s ~ n_sp_missing_k, data = .))) |>
# dplyr::ungroup()
ggplot(speed_out, aes(x = n_sp_missing_k, y = time_s, color = expr, group = expr)) +
geom_point() +
geom_smooth(method = "lm") +
labs(x = "Number of binding species (Thousand)",
y = "Time (Second)", color = NULL, group = NULL) +
scale_x_continuous(breaks = c(0.05, seq(0.5, 5, 0.5))) +
scale_y_continuous(breaks = c(0, 10, seq(25, 200, 25))) +
geom_text(x = 4, y = 30, label = "rtrees: y = 0.35+ 0.95x", inherit.aes = FALSE, color = "#F8766D") +
geom_text(x = 1.8, y = 150, label = "V.PhyloMaker2: y = 12.3 + 36.8x", inherit.aes = FALSE, color = "#00BFC4") +
theme(legend.position = "none")
#> `geom_smooth()` using formula = 'y ~ x'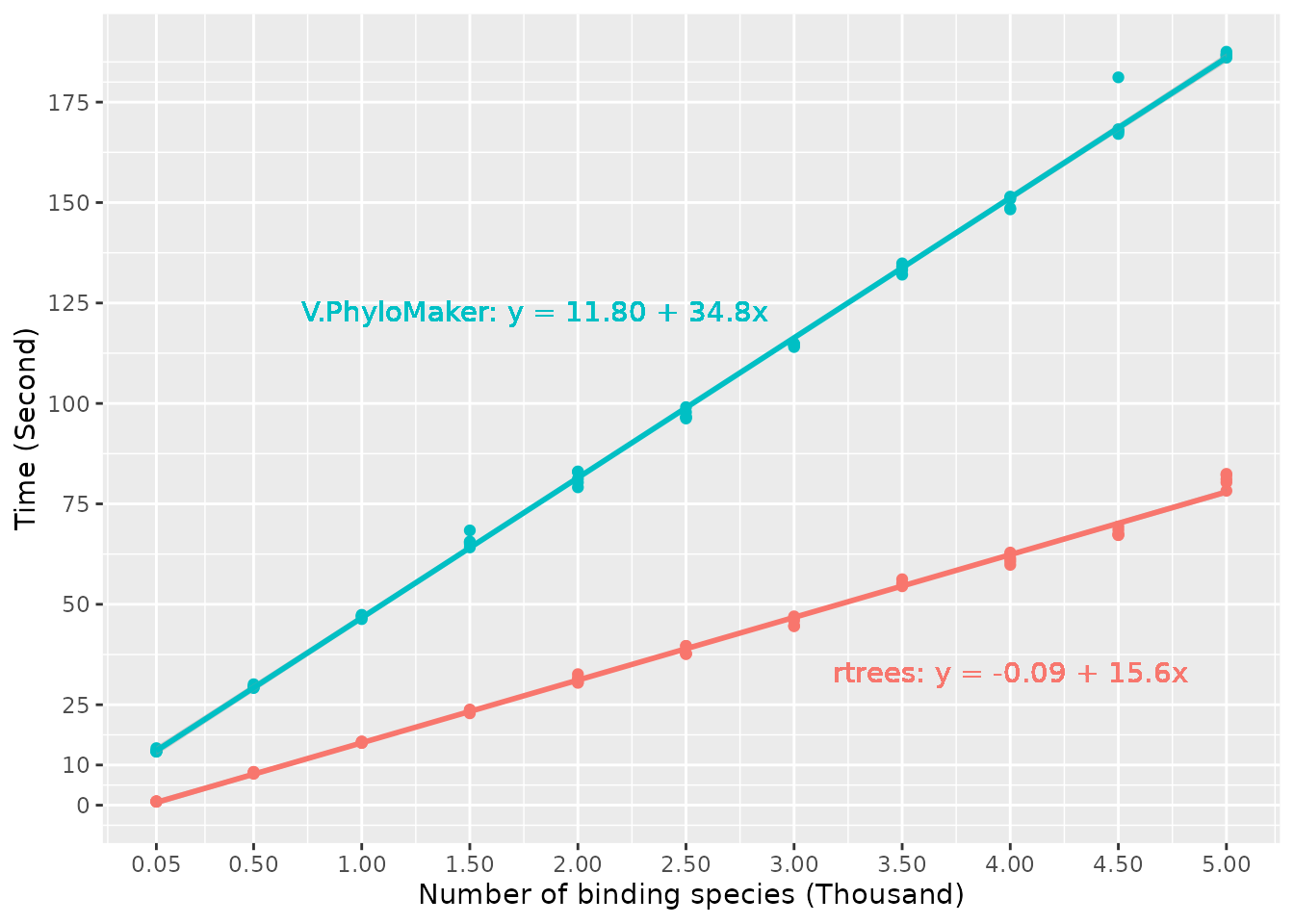
Based on the figure above, the time used by rtrees (and
other R packages such as V.PhyloMaker and
FishPhyloMaker) increased linearly with the number of
missing species. This is because of the sequential structure of the R
phylo object, in which all tips and nodes of a phylogeny
are labeled with continued integers. When a new node or tip needs to be
inserted, all the numbers after the binding location need to be pushed
one place after. Therefore, the binding of tips in an R
phylo object is a sequential process, which cannot be
conducted with parallel computing to take advantage of the multiple CPU
cores a regular computer has nowadays. It is possible to speed it up by
converting some core R code into C++. However, it takes a lot of effort
to do this right.
Another potential resolution is to represent phylogenies in a
non-sequential structure unlike phylo objects in R. One
example is the treeman
package, which developed such a structure to perform tree
manipulations. Because of the non-sequential structure (i.e., no
continuous labeling of nodes and tips), it is possible to use parallel
computing to process phylogenies. However, such a structure has not yet
been adapted widely by the R community. Therefore, the infrastructure to
support such a non-sequential structure in R is still missing, making it
hard to use it here in rtrees.
Despite the limitations described above, rtrees is
reasonably fast and user-friendly with informative messages.